How often should I have a screening mammogram?
As most women know, for the last several years the answer to this question has been steeped in controversy and confusion. For decades, it was recommended that annual screening be carried out starting at age forty. But, starting with a report from the United States Preventive Services Task Force (USPSTF) in 2009, and subsequently, in 2011, from the Canadian Task Force on Preventive Health Care (CTFPHC), biennial mammograms were felt to be sufficient for women ages 50 to 74 at "average risk" for developing breast cancer; for all others, annual mammograms were recommended.
Those task force guidelines notwithstanding, there is still an ongoing controversy about the optimal screening interval for average-risk women, with the American Cancer Society still recommending annual screening for women between 45 and 54, but adopting the USPSTF approach for women of 55 and older, while the National Comprehensive Cancer Network (NCCN) continues to recommend annual mammograms for all women "starting at age 40 for as long as a woman is in good health". In Canada, most provincial screening programs have adopted the two-year interval for women at average risk, while the U.K. National Health Service recommends every three years for average-risk women ages 47 to 73. However, it is important to understand that in all these jurisdictions, a woman can still opt to have a yearly mammogram ordered by her physician outside the national or provincial screening programs.
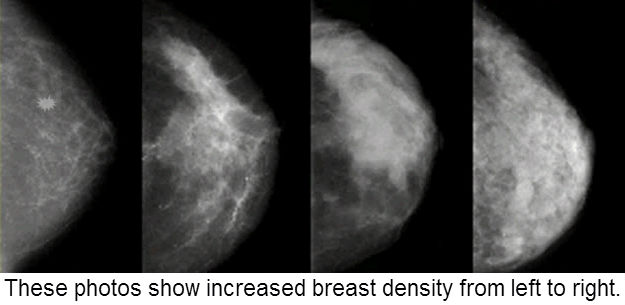
A major drawback to establishing broad screening guidelines is trying to make one size fit all within large populations. Now, in a just-published study in the Annals of Internal Medicine, researchers from the Cancer Intervention and Surveillance Modeling Network, collaborating with the Breast Cancer Surveillance Consortium (BCSC), suggest that for women ages 50 to 75, screening intervals should be based on a combination of breast density (a radiological finding on the mammogram, seen in the images at the top of this blog) and a woman's other risk factors for breast cancer (such as a family history), an approach that may be better tailored to each individual.
The authors of this study recommend that, for women with low breast density on mammograms and low to average risk factors for breast cancer, screening every 3 years may be sufficient, while for women with radiologically-dense breasts and/or other risk factors, yearly screening should be undertaken.
Having radiologically-dense breast tissue on mammograms has been linked to an increased risk of developing breast cancer coupled with greater difficulty in detecting tumors when they are small because the dense breast tissue often obscures them.
That said, what else besides a family history puts a woman in the high-risk category?
- Obesity (greater than 20% over ideal body weight)
- Enlarged thyroid (including Hashimoto’s thyroiditis)
- Diabetes
- Never having been pregnant (nulliparity)
- First pregnancy age 32+
- Regular alcohol (all types) consumption starting in teenage years
- Smoking, including chronic exposure to second-hand smoke
- Prolonged use of oral contraceptives during reproductive years
- Hormone replacement therapy (HRT) after menopause




 RSS Feed
RSS Feed
